INTRODUCTION
The non-alcoholic fatty pancreatic disease (NAFPD) consists in the excessive fatty accumulation of the pancreas, and the pancreatic steatosis is defined as fatty infiltration in the pancreatic islets or acinar cells(1), as consequence of ectopic fat deposition in a non-fatty tissue. Studies estimate the prevalence of NAFPD to be 10% in the pediatric population and to range from 16 to 35% in that in the adult population(2). A high frequency between NAFPD and type 2 diabetes mellitus (DM2) has been observed(3). Emphasis has been placed on the influence of pancreatic steatosis as a cause of beta-cell dedifferentiation due to increased fatty acids in the pancreatic islets, this being the likely key mechanism for the onset of DM2(4).
The beta-cell dedifferentiation is defined as a phenotypic alteration of the beta-cell that leads to the loss of its insulin-secreting function and consequent triggering of diabetes mellitus through negative gene regulation of beta-cell, or the simultaneous positive regulation of genes expressed or suppressed in normal beta-cell, or the possible positive regulation of genes from progenitor cells(5). However, it can also correspond to damage of differentiated cellular elements, leading to heterogeneity of immature primitive beta-cell(6). Thus, the beta-cell dedifferentiation ultimately leads to a major dysfunction in insulin secretion.
Beta-cell dedifferentiation as a mechanism of beta-cell failure and development of DM2 was a concept introduced Talchai et al(7). Probably, the indirect effects associated with insulin resistance (IR) have an important impact on beta-cell dedifferentiation(8). The beta-cell mechanisms that protect against dedifferentiation have not yet been determined.
Studies show that NAFPD is associated with IR(9). The IR increases during the course of DM2 progression; however, beta-cell function deteriorates rapidly, and this functional depletion will distinguish individuals who will develop beta-cell dedifferentiation evolving to diabetes or not(10).
The purpose of this study was to evaluate the interaction between NAFPD and IR as a mechanism of beta‐cell dedifferentiation in development of DM2 through the signaling pathway design, based in research articles.
MATERIALS AND METHODOS
Modeling
The design of molecular pathway maps implies careful extraction of molecular characteristics from the literature and other means accompanied by setting of the multiple elements into a network of interconnected and interacting events.
Based in research articles, we selected well-documented pathways and obtained specific expression profiles of these pathways. PubMed and Google Scholar were utilized to search for proofread publications that have investigated the NAFPD, IR, and beta‐cell dedifferentiation. The signaling pathway reference data was selected by incorporating information from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The KEGG is a database with a collection of biological information about signaling pathway maps, genome sequencing, biological process networks, protein domains, with the goal of utilizing and understanding these functions at a high level(11).
We investigated a total of 30 signaling pathways in the KEGG databases, and for each pathway we identify all the proteins related to pancreas, beta-cell, IR, and DM2 commons between beta‐cell dedifferentiation and NAFPD. We subsequently compared the proteins and the six-way interactions. The KEGG database includes various types of information about proteins, containing pathway reports demonstrated in graphical models that make it possible to conceive of protein interactions in complex biological methods.
The MODELLER 10.1 package was used to predict 3-dimensional structure based on the homology modeling protocol. MODELLER is software for comparative modeling of protein structure by alignment of a sequence that will be modeled with the model structures. Based on the resulting structure, we have designed the peptide activators of signaling pathway.
The signaling pathway diagram design was done with PathVisio software (version 3.2.4), and was utilized for the graphic demonstration of signaling pathway since it is a tool that allows view and edit of biological signaling pathways. PathVisio is a public domain pathway editor, analysis and visualization software that is incorporated into the community pathway database WikiPathways one popular free of charge accessible databases for biological pathways evaluation. However, our study was conducted using the KEGG database.
RESULTS and DISCUSSION
Non-alcoholic fatty pancreatic disease, insulin resistance, and beta‐cell dedifferentiation – Signaling pathway diagram design
In our work, we coupled a review study with computational modeling to understand the signaling mechanisms that lead NAFPD to trigger IR and beta-cell dedifferentiation having DM2 as an outcome.
The several basic mechanisms of NAFPD as well as their interaction with beta‐cell dedifferentiation in triggering of DM2 are demonstrated in Figure 1.
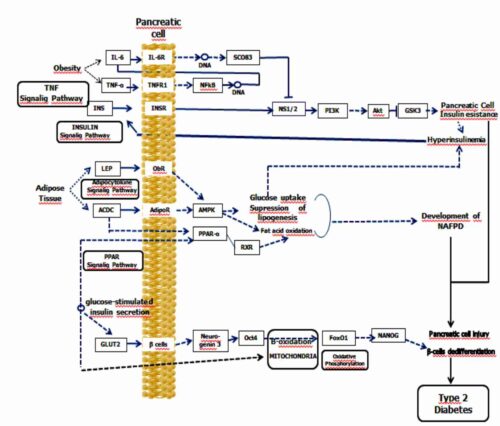
Figure 1. Non-alcoholic fatty pancreatic disease, insulin resistance, and beta‐cell dedifferentiation – Signaling pathway diagram design
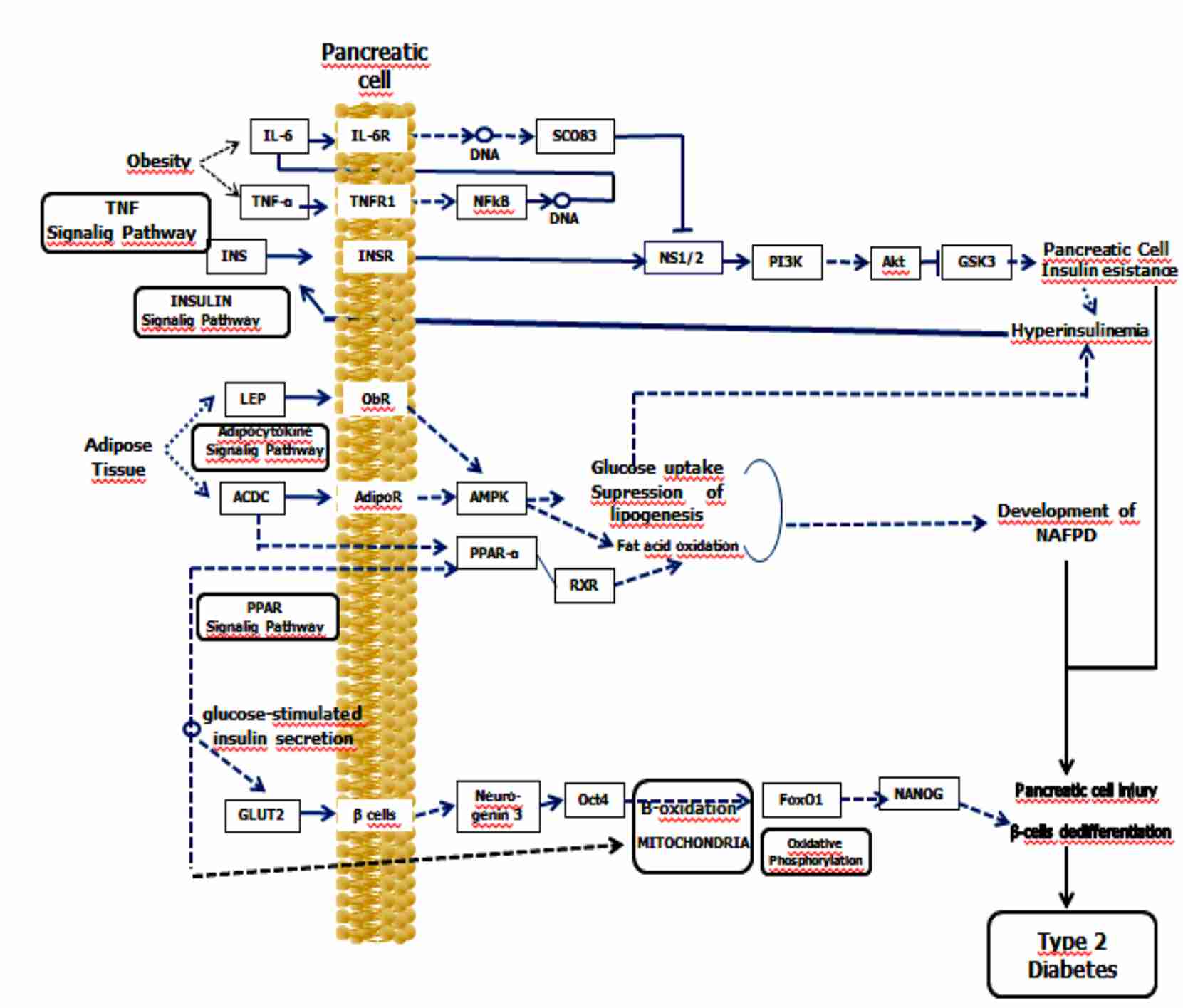
NAFPD describes a spectrum ranging from simple pancreatic steatosis to severe steatopancreatitis with pancreatic inflammation and fibrosis, referred to as non-alcoholic steatopancreatitis (NASP).
In the early phase of NAFPD an accumulation of adipose tissue occurs activating inflammatory factors. The activation of interleukins is primarily caused by the IR, leading to altered insulin release and elimination of free fatty acids (FFA)(12). In addition, the activation of a key-enzyme of the lipogenesis by factor Peroxisome Proliferator-Activated Receptor-α (PPAR-α) leads to increased FFA synthesis by the pancreas(13). As NASP progresses into a subsequent phase, as a result of increased endoplasmic reticulum stress and oxidative stress via mitochondrial beta-oxidation, an increase in reactive oxygen species occurs with consequent lipid peroxidation. This lipid peroxidation increases the production of cytokines and caspases leading to apoptosis, inflammation, and fibrosis(14). Chronic inflammation is one of the causes leading to IR, and the inflammatory state subsequent to visceral fat increment is one of the determinants of pro-inflammatory cytokine production, comprising interleukin 6 (IL-6), tumor necrosis factor alpha (TNF-α), and the profibrogenic adipokine leptin. Furthermore, progression to IR following deactivation of the regulatory signaling pathway→serine/threonine kinase (STK) phosphoinositide-3-kinase (PIK3), leads to the development of IR of pancreatic cells resulting in NAFPD.
Several mechanisms are involved in the genesis of IR, comprising incrementation of insulin receptor substrate (IRS) protein phosphorylation by the inhibitor of nuclear factor kappa B kinase subunit beta, c-Jun N-terminal protein kinase 1, and protein kinase C(15). The IR has with consequence also deterioration of the IRS-1 proteasome through mammalian target of rapamycin (mTOR), elevating the effects of phosphatase and tensin homolog (PTEN), of protein phosphatase 2A (PPA2) and of protein tyrosine phosphatase (PTPs); furthermore, it reduces the activation of the signaling molecules such as protein kinase B (AKT) and PI3K(16).
Cell differentiation involves synchronous and tightly controlled activation/repression of specific genes and effectors in a time-dependent manner, giving rise to cells with specific morpho-functional properties. Dedifferentiation, on the other hand, involves the loss to varying degrees of cellular identity and phenotype and may regress to a less differentiated or precursor-like condition(5).
Recent data support that beta-cell dedifferentiation alters insulin secretion, and plays an important role in triggering DM2(7,17). Thus, the beta-cell dedifferentiated has a tendency to transform into other non-beta cell endocrine cells that cause the development of diabetes(18).
The disturbance in glucose metabolism involves high demand for insulin secretion with increased levels of reactive oxygen species (ROS) in the beta-cell that consequently release chemokines that recruit macrophages leading to increased beta-cell dedifferentiation by inflammatory processes with reduced beta-cell mass(8). The beta-cell are especially sensitive to oxidative stress because of their elevated ROS production with reduced antioxidant potency, indicating that oxidative stress may play a significant role in pancreatic beta cell failure resulting in DM2(19).
Glucose transporter 2 (GLUT2) is main glucose transporter in beta-cells of pancreatic islets, mediating the diffusion of glucose through of cell membrane, and plays a crucial role in glucose metabolism(20).
Neurogenin 3 is a pancreatic endocrine lineage-specific marker being an important regulator in beta-cell regeneration and differentiation, as well as stimulating the expression glucose sensor glucokinase of beta-cell. However, the neurogenin 3 does not stimulate the glucose transporter GLUT-2(21,22).
The octamer-binding transcription factor 4 (Oct-4) is a protein that is implicated in maintaining the self-renewal of undifferentiated, pluripotent embryonic stem-cells. There are three isoforms of Oct-4, of which Oct-4b is essential in the response to stress, whereas the other isoforms are related to plasticity and to the activation or inactivation of beta-cell dedifferentiation(23).
Mitochondrial beta-oxidation involves linear chain fatty acids structured into two functional subdomains by via a complex pathway under intramitochondrial command. Increased fatty acid beta-oxidation reduces lipid accumulation in the cytoplasm by decreasing glucose metabolism, and incomplete oxidation potentially contributes to IR(24). In vitro studies demonstrate that high levels of fatty acids induce glicotoxicity and beta-cell dysfunction; thus, lipid accumulation in the pancreas demonstrated the presence of IR and DM2(25).
Forkhead box protein O1 (FoxO1) is essential for insulin action. FoxO1 is a conserved transcription factor that has direct interaction with genes involved in gluconeogenesis and metabolic regulation(26). FoxO1 regulates glucose homeostasis in beta-cells and peripheral tissues, and its activation leads to IR and beta-cell dedifferentiation, while decreasing its function can restore pancreatic beta-cell function by reversing hyperglycemia(27).
Oxidative phosphorylation in mitochondria is made up of five complexes that are fundamental in regulating cellular metabolism, and precisely the ATP-producing oxidative phosphorylation pathway is essential in insulin secretion.(28) Recent studies put altered oxidative phosphorylation as a key genetic component of IR(29).
Nanog is as one of the heterogeneously express genes in induced pluripotent stem-cell and embryonic-stem populations. It performs an essential role in self-renewal cellular, maintenance, and pluripotency. In addition, Nanog has directed each other interactions with Oct4 and Sox2 genes(30). The pluripotent bone marrow stromal cells that co-expressing NANOG can be differentiated in beta-cells(31). Beta-cell dedifferentiation happens often in DM2 and is link to with an arising loss of FoxO1 function, and there exists an opposite link between beta-cell differentiation and FoxO1 as DM2 progresses, with a significant elevation of Nanog expression(7).
CONCLUSIONS
Beta-cell dedifferentiation happens often in DM2, and the interaction between NAFPD and IR prove to be two important indices to the possible mechanism of beta‐cell dedifferentiation in development of DM2 as demonstrated through in signaling pathway.
CONFLICT OF INTEREST: The authors declare that there is no conflict of interests regarding the publication of this paper.